Medical Device Regulation (MDR) Compliance Services
The EU Medical Device Regulation (MDR 2017/745) has replaced the Medical Device Directive (MDD). It imposes stricter requirements on manufacturers to ensure the safety, quality, and performance of medical devices marketed in the European Union.
What is MDR?
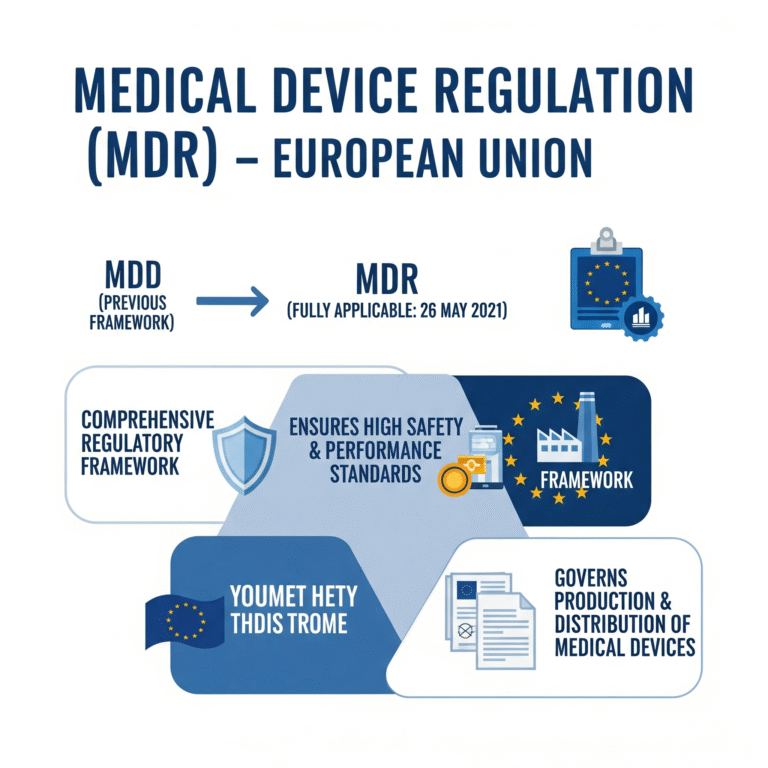
The Medical Device Regulation (MDR) is a comprehensive regulatory framework enforced across the European Union. It governs the production and distribution of medical devices, ensuring they meet high safety and performance standards. MDR became fully applicable on 26 May 2021, replacing the previous MDD.
Key elements of MDR include:
- Increased clinical evidence and post-market surveillance
- More detailed technical documentation
- Strict requirements for high-risk devices
- Greater transparency through the EUDAMED database
- Appointment of a responsible person (PRRC)
Why is MDR Compliance Important?
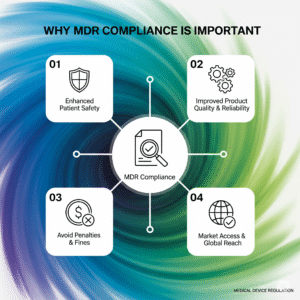
MDR compliance is mandatory for all manufacturers selling medical devices in the EU. It:
- Ensures legal market access within the EU
- Demonstrates commitment to safety and quality
- Prevents legal liabilities and product recalls
- Increases transparency and strengthens consumer confidence
Our MDR Compliance Services
Lisora helps your organization transition and comply with MDR requirements efficiently and confidently. Our services include:
- Gap analysis and transition planning from MDD to MDR
- Technical documentation creation and updates
- Clinical evaluation report (CER) preparation
- Post-market surveillance planning
- EUDAMED registration and PRRC support
Each device class has specific regulatory requirements based on its risk level.

Why Choose Lisora for MDR Compliance?
MDR Specialists
In-depth knowledge of EU regulatory rules worldwide
Regulatory Alignment
Ensure your QMS meets ISO 13485 & MDR standards
End-to-End Support
From initial check to notified body submissions fast
Timely Execution
Reduce time-to-market and cut audit risks & quickly
Frequently Asked Questions (FAQs)
1. What is the MDR regulation?
MDR (EU 2017/745) is the European Union’s regulation for medical devices that ensures safety, efficacy, and transparency.
2. Who must comply with MDR?
All medical device manufacturers, authorized representatives, importers, and distributors intending to sell in the EU must comply with MDR.
3. Is MDR applicable to existing CE-marked devices?
Yes. Legacy devices CE-marked under MDD must transition to MDR before the applicable deadlines or they cannot be sold in the EU.
4. What is the role of PRRC in MDR?
The Person Responsible for Regulatory Compliance (PRRC) ensures that the device manufacturer meets all regulatory obligations under MDR.
5. What is EUDAMED and why is it important?
EUDAMED is the European database for medical devices that increases transparency and traceability throughout the device lifecycle.